Пикотехнология позволяет конструировать молекулярные механизмы на электронном уровне с точностью долей пикометра.
Согласно современным научным данным, наномашины - это механизмы из атомов, сконструированные с точностью нанометр. Сложные наномашины, в частности белки, создаются при помощи живой клетки. Искусственные белки, изготовленные без помощи живой клетки, точнее без помощи рибосомы (белковой фабрики клетки), примитивны по сравнению с натуральными. Посмотрим, с чем это связано.
Атомы, объединяясь в молекулы, проявляют "странные" квантовые свойства. Углы между связями атомов в молекулах часто похожи на углы между уложенными вплотную многогранниками.
Оказалось, что и поворачиваться вокруг химических связей атомы могут чаще всего дискретно, как будто грани атомов смагничиваются с гранями соседних атомов. Специалисты по квантовой химии, молекулярной биологии и кристаллографии очень удивились, когда мы показали им модели известных молекул и кристаллов, где многогранные атомы действительно смагничены своими гранями.
В наших моделях гранями атомов являются кольца-электроны, магнитные свойства которых измерены с большой точностью экспериментально. Кольцевая форма нашей модели электрона позволяет в модельном эксперименте продемонстрировать устойчивость 1,2,8,18,32-гранных оболочек атома и, таким образом, понять процесс формирования электронной структуры атома и определяемые им периодические особенности, отраженные в таблице Менделеева.
Это уже другая смежная область пикотехнологии, названная нами атомной геометрией. Пикотехнология ставит целью адекватно представить механизм молекулы на электронном уровне. Что это значит? Если наша кольцевая модель электрона правильно отражает реальный электрон, то сконструированные из колец модели электронных поверхностей должны иметь форму и независимые от масштаба свойства реальных молекул. Мы занялись этой проверкой и обнаружили, что кроме геометрической формы наши модели позволяют увидеть причины многих "странных" свойств молекул. Почему, например, в аминокислотах атомы пептидной группы ведут себя подобно локтевому суставу?(в плоскости сустава молекула более гибкая, чем в перпендикулярной плоскости). Взглянув на "электронный механизм" легко понять, почему он обладает свойствами сустава.
Кольца-электроны могут свободно кататься друг по другу, но не могут деформироваться. Однако, достаточно расположить восемь колец симметрично и мы получим жесткую конструкцию восьмигранного атома.
Так, все же, как нам наглядно убедиться, что формы моделей из колец точно совпадают с формами реальных молекул? Это не так просто, как может показаться на первый взгляд. Дело в том, что не только электроны, но и многие атомы на электронных фотографиях молекул неразличимы. Общий контур молекулы хорошо различим лишь для крупных белков, насчитывающих тысячи и более атомов. Мы решились построить кольцевые модели нескольких известных белковых молекул. Одна из них troponin-C, насчитывающая около 8 000 поверхностных электронов. Она построена не по результатам рентгеноструктурного анализа, а по генетическому коду этой молекулы.
В современных учебниках по биологии написано, что белковые молекулы рождаются в развернутом виде, а затем сами сворачиваются под действием межатомных сил. Однако, в пробирке этого может и не произойти. Почему? Мы построили модель рибосомы, которая собирает белки по коду и обнаружили, что присоединение аминокислоты может происходить под разными углами (три кольцевых грани одной аминокислоты могут быть примагничены к трем кольцевым граням другой аминокислоты тремя способами).
Угол поворота аминокислоты кодируется третьей буквой триплета ДНК. Расшифровав композиционный генетический код (эта таблица опубликована в газете "Экономика сегодня и завтра №2(4) за 1993 г.) мы смогли вручную, а затем на компьютере строить по коду пространственную структуру любого белка.
Компьютерная программа "Пикотехнология", созданная по нашему проекту фирмой "А-ГРАФ", является достойным дополнением международной программы "Геном человека", цель которой - определение генетической последовательности всех хромосом человека.
По кодам, определенным программой "Геном человека" наша программа "Пикотехнология" сможет построить 100 000 пространственных структур белков, в том числе 2 000 известных человечеству сегодня. Построение одной структуры длится сотые доли секунды. Интересно, однако, продемонстрировать процесс сборки белковой молекулы по генетическому коду. Хитрость, которой воспользовалась природа для кодирования формы белков, заключена в стандартной форме пептидной группы всех аминокислот.
Специалисты с недоумением отмечают, что замена многих аминокислот практически не влияет на форму и функции белковой молекулы. С нашей точки зрения это не удивительно, т.к. стебель белка, состоящий из пептидных групп остается той же формы.
Мы рассмотрим конструирование только стебля белковой молекулы. Вы увидите, что это достаточно для получения пространственной структуры любого белка, воспроизводимого рибосомой за один проход. Аминокислота, сборочная единица белковой молекулы, упрощенно выглядит следующим образом: Представьте себе винтовую лестницу с перилами. Нас будут интересовать только перила, точнее одно из них. Оно похоже на гигантский штопор, не правда ли ? Вырежем из этой формы участок, описывающий в проекции дугу в 90 градусов, и модель готова. Сечение нашего гигантского штопора должно быть треугольным, причем, треугольники (АBC и DEF) должны быть правильными.
Каждый следующий участок белка (аминокислота) присоединяется к предыдущему так, чтобы треугольник ABC следующей аминокислоты совпал с треугольником DEF предыдущей. Сколько вариантов присоединения? Правильно, три. Каждый из трех вариантов имеет свой генетический код (третья буква триплета). Например, аминокислота глицин Giy кодируется кодом GGC, если входит в состав альфа-спирали, кодом GGA, если входит в состав бета-слоя, кодом GGG, если входит в состав уменьшенной альфа-спирали, где водородная связь установлена не с четвертым, а с третьим аминокислотным остатком и, наконец кодом GGT, если является вторым аминокислотным остатком бетаповорота. Итак, третья буква триплета указывает под каким углом нужно примагнитить следующую пептидную группу к предыдущей.
Механизм композиционного кодирования
1). Вариант альфа. Последовательное присоединение аминокислот под таким углом формирует альфа-спираль белка.
2). Вариант бета. Последовательное присоединение аминокислот под углом бета формирует бета-слой.
3). Вариант кси. Образует, так называемые, петли. Последовательность -альфа-кси-альфа- позволяет сделать поворот белковой цепи в бета-слое.
Вот, собственно и вся азбука белковой архитектуры. Комбинация вариантов альфа, бета, кси позволяет воспроизвести невообразимо сложные формы белковых молекул. Нужно, конечно отметить, что реальные белки взаимодействуют еще и посредством радикалов аминокислот, которые могут выполнять функции магнитных защелок, демпферов, шарниров, суставов, штампов, замков и ключей, щупалец и рук, словом всех невообразимо сложных функций привычного нам мира и пока непривычного нам микромира.
Участки белков, воспроизводимых рибосомой за один проход, могут собираться посредством ферментов в сложные конгломераты четвертичной структуры, из которых в дальнейшем могут быть образованы структуры органелл(органов живой клетки). Но наша тема связана лишь с азбукой архитектуры молекул.
С этой точки зрения рассмотрим модель механизма ДНК, образованную как бы из двух "живых" молекул РНК со снятыми ограничителями, выполняющими в РНК функцию позвонков. Какие ограничители, какие позвонки в молекуле? Да, представьте себе, что функционально молекула РНК похожа на позвоночное животное. У нее есть "позвоночник", состоящий из "молекул-позвонков" фосфорной кислоты, которые, согласно нашей модели электрона и атома, функционально похожи на кубики Рубика.
Атомы в них могут поворачиваться, но магнитные свойства электронов удерживают их в трех устойчивых положениях, фиксируя разные устойчивые формы РНК и ДНК.
У этого "позвоночного животного" есть "магнитная чешуя", которая может образовать ступеньки спиральной лестницы, состоящей из двух "позвоночных животных". Головная "чешуйка" может держать четырьмя магнитными "присосками" аминокислоту и под нужным углом "вмагнитить" ее в создаваемую молекулу белка. В конце концов, она плавает как живая. Может быть она и на самом деле живая? Ведь что такое жизнь? Не будем касаться разумной ее формы. Жизнь - это прежде всего самовоспроизведение. Магнитные чешуйки нашего "позвоночного животного" позволяют ему размножаться (при наличии условий конечно).
Условия могут воспроизводиться по программе, записанной на магнитных "чешуйках". Для выполнения этой программы могут потребоваться сложные механизмы белковых молекул, клетки, человека, государства, цивилизации... Но вернемся к азбуке. Вопрос первый: как и почему может плыть наше "позвоночное животное?"
Его кольцегранная "шкурка" обладает необычным для нашего мира свойством. Кольца-электроны образуют "скользкую" поверхность, "кожу молекулы". В некоторых кольцах снаружи удерживаются ядра атомов водорода, протоны. Их размер в сто тысяч раз меньше кольца-электрона. "Кожа молекулы" обладает как бы шипами, на которые наскакивают в своем тепловом движении мелкие молекулы воды и отдельные атомы и ионы.
Если мы посмотрим на модель РНК, то увидим, что "шипы" направлены в одну сторону. Таким образом, мелкие молекулы, ударяя по РНК со стороны "головы" проскальзывают, а со стороны "хвоста" натыкаются на "шипы" и подталкивают РНК вперед. Так в теплой воде "оживают" молекулы РНК.
Вопрос второй: "как построить подобную себе молекулу?" А) Доплыть до "супа позвонков с чешуйками" Б) Подождать, пока "чешуйки" примагнитятся, синхронно образуя "позвоночник" информационной копии. В) Подождать "весны", когда температура среды повысится на столько, что "спаренное позвоночное животное" "расплавится" на две "особи". Вопрос третий: "как приготовить "суп" и не ждать "весны"?" А) Развить генетическую программу до уровня самосознания. Б) Прочитать кулинарную книгу.
К сожалению в популярную статью не вместить всех нюансов живой архитектуры, поэтому массу вопросов по пикотехнологии, которые должны появиться у читателей, можно выяснить, посетив наши лаборатории "Наномир" и "Пикотехнология". Постоянная выставка, насчитывающая 1 500 форм кольцегранных атомов, молекул, кристаллов и фракталов, находится в центральном павильоне ВВЦ.
Пока статья готовилась к публикации нам удалось получить экспериментальное подтверждение работоспособности нашей таблицы композиционного генетического кода, поэтому мы приводим ее полностью для тех, кто хочет увидеть форму любого белка, имея лишь его генетический код.
Для простоты преобразования "код аминокислоты - композиция" триплетный генетический код заменим номерами от 1 до 64 по которым из нашей композиционной таблицы будем определять вариант композиции. Вариант С означает альфа-спиральный участок, А - бета, Т - композиция второго остатка бета-поворота, G - уменьшенная альфа-спираль.
Таблица композиционного генетического кода:
TCCTACGTTCCTACCTTACCACGTACGTACGTTCCTACGTACGTACGTCCCTACGTCACCTCCT
Посмотрим, как определить композицию аминокислотного остатка Gly по его генетическому коду GGC.
1. Превратим код GGC в номер, учитывая, что A=0, C=1, G=2, T=3 в четверичной системе счисления. GGC=16·2+4·2+1·1=41.
2. Отсчитываем в таблице композиционного кода 41-й символ (обратите внимание, что таблица начинается не с 1-го, а с нулевого номера). Итак, если Вы не ошиблись, то найденный символ - это символ С, что означает, что наш глицин входит в состав альфа-спирали. Кому интересно узнать структуру белка по коду, а вручную считать утомительно, предлагаем воспользоваться нашей компьютерной программой. Программа написана на стандартном Паскале и позволяет преобразовать генетический код белка в композиционный код его структуры, согласно нашей таблице.
А теперь посмотрим, что получится после преобразования генетического кода альфа-структурного белка керотина и бетаструктуроного белка фиброина. Первые три строчки - результат работы нашей таблицы над кодами альфа-керотина, а следующие три строчки - бета-фиброина.
Каждый, кто отличит букву C от T увидит, что первые строки, в отличие от последующих, содержат сплошные С, означающие альфа-структуру.
См. также научную статью в журнал "Биохимия"
http://nanoworld88.narod.ru/data/297.htm
Международный конкурс "Пикотехнология-2012"
Кушелев: А что нам мешает начать собственный международный конкурс по определению структуры белков? В отличие от организаторов CASP10 мы не будем засекречивать список участников и данные, опубликованные каждым из них на форуме лаборатории Наномир.

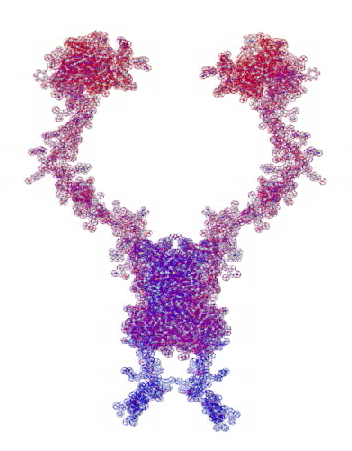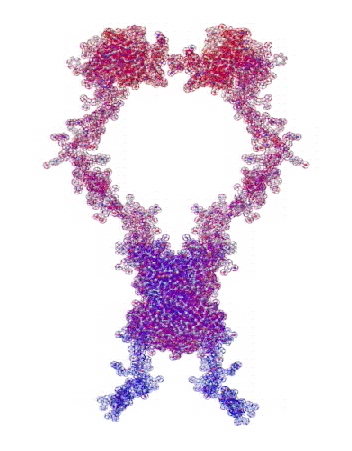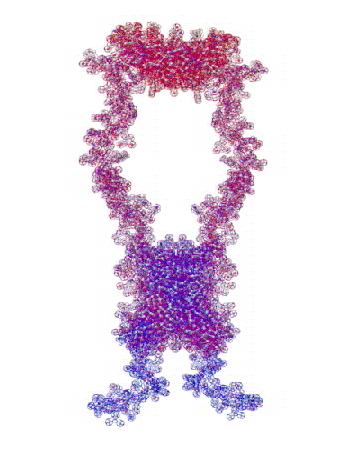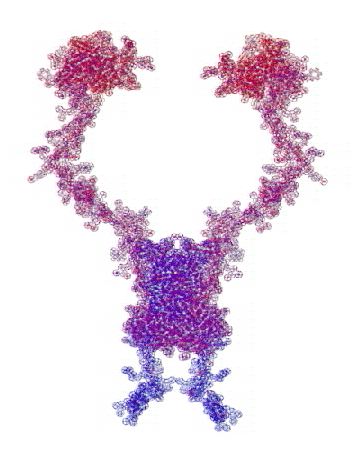


На конкурсе "Пикотехнология-2012" каждый может публиковать не только pdb-файлы белков, в т.ч. с конкурса CASP10, но и любые другие модели атомов, органических и неорганических молекул и кристаллов, витаминов, ДНК/РНК и т.д.

Пикотехнологическая модель несопряжённой связи сопряжённых систем. Орто-дитриазин.
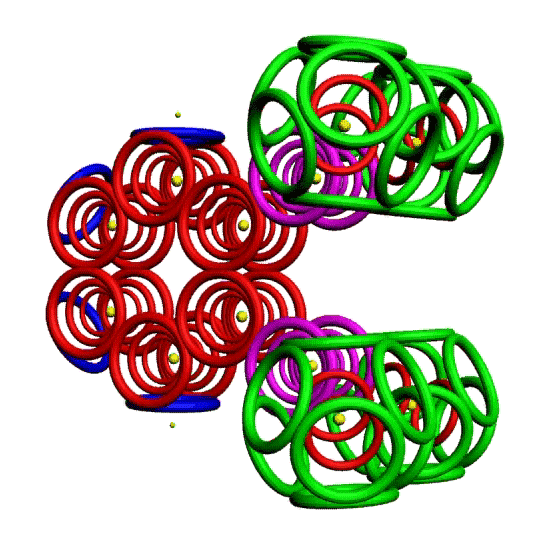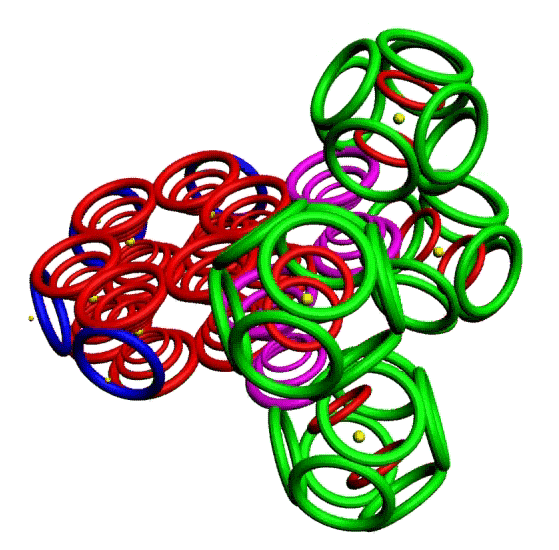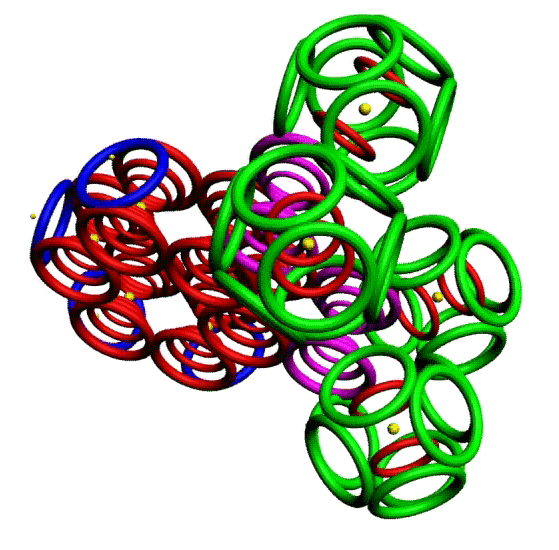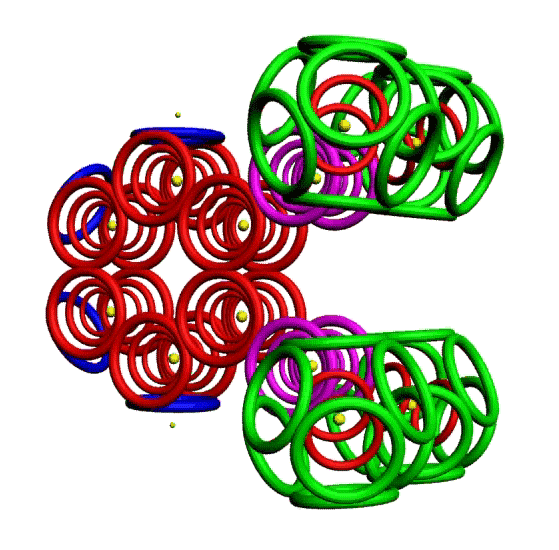

m-Динитробензол o-Динитробензол Гексанитробензол
Материал с форума лаборатории Наномир:

Kushelev: ... а для молекулярного иона HCNH+ ...
Игорь: Нету такого иона: есть лишь CN- (с минусом).
Кушелев: Случайно обнаружил, что ион HCNH+ всё-таки существует, и о нём написано всё в той же Википедии: http://en.wikipedia.org/wiki/HCNH%2B
Цитата: The ground state structure of HCNH+ is a simple linear molecule. In addition, there are multiple higher energy isomers such as CNH2+, H2CN+, cis -HCNH+, and trans -HCNH+. [1] Конец цитаты.

В ~1992-ом году я сделал пикотехнологическую модель молекулы NO, общая молекулярная поверхность которой образована 6+5=11 электронами кислорода и азота соответственно. Эта же модель подходит и для молекулярного иона HCNH+, причём позволяет смоделировать не только линейную молекулу, но так же транс- и цис-изомеры... Есть и любопытный нюанс. Транс-изомеров получается два. Правый транс-изомер и левый транс-изомер. А это уже можно проверить по вращению поляризации света...
Кстати, имея две модели линейного иона HCNH+ можно предположить о существовании нового типа изомеров, один из которых состоит из многогранных атомов, а другой из многослойных. Такого типа изомеры имеет смысл назвать новым термином, например, Ку-изомеры![]()
От обычных изомеров они отличаются принципиально. Например, ядра обоих Ку-изомеров могут лежать на одной прямой, но при этом находиться на разных расстояниях друг от друга. Кстати, мы фактически столкнулись с аллотропией электронных оболочек атома/молекулы.
ПИКОТЕХНОЛОГИЯ – НОВЫЙ ПОДХОД В МОДЕЛИРОВАНИИ
ПРОСТРАНСТВЕННОЙ СТРУКТУРЫ БЕЛКА
Кушелев А.Ю., Соколик В.В.
Научно-исследовательская лаборатория "Наномир", Дмитров
ГУ «Институт неврологии, психиатрии и наркологии АМНУ», Харьков
В настоящее время наибольшей популярностью пользуются научные исследования с приставкой «нано»: речь идёт об объектах, размер которых лежит в диапазоне 1-100 нанометров [3, с. 28; 16, c. 6]. Однако при моделировании атомов в составе биомолекул возникла необходимость оперировать конфигурацией электронов, формирующих внешний валентный уровень, который и определяет объем, занимаемый тем или иным атомом в пространстве, а это уже размерность пикометров (10-12 м). Пикотехнология, методический подход для моделирования пространственной структуры белка, опирается на следующие представления о микромире:
· кольцегранная структура электронных орбиталей (электронов) в атоме, которая детерминирует в первом приближении форму атома в виде усечённого октаэдра (рис. 1), грани которого – электроны валентного энергетического уровня [17, c. 49; 4, с. 46; 1, с. 73];
· геометрический алгоритм объединения атомов в молекулах определяет не только межатомные расстояния (длины связей), но прежде всего углы поворота по этим связям, кратные 120о в трёхмерном пространстве [2, c. 237; 5, с. 137];
· ротамерный вариант пептидной связи, которая объединяет аминокислотные остатки в полипептид, детерминирован третьим нуклеотидом кодона [2, c. 237; 18, c. 347] и реализуется в ходе матричного синтеза структурного шаблона белка на рибосоме пулом изоакцепторных тРНК [6, c. 13].
А Б

Рис. 1. Кольцегранная модель валентной электронной оболочки атома из 8 электронов (А) и её аппроксимация усеченным октаэдром (Б) в моделях белков.
Десять лет назад А.Ю. Кушелевым была сформулирована идея композиционного генетического кода [2, c. 237], которая была расширена представлениями о кодировании ротамерии пептидной связи и структурного шаблона белка [18, c. 348; 19, с. 275; 20, c. 118]. В таблице композиционного генетического кода каждому варианту композиционного кода, в зависимости от кодона, соответствовало определённое значение композиционного угла, под которым в ходе матричного синтеза происходит присоединение очередного аминокислотного остатка к растущей полипептидной цепи. Данная таблица легла в основу алгоритма первого варианта компьютерной программы «Пикотехнология» для моделирования пространственной структуры белка по детерминирующей его нуклеотидной последовательности. Дальнейшая разработка данной проблемы привела к уточнению не только самой таблицы, но и к введению таких понятий, как ротамерия пептидной связи и структурный шаблон белка. Было установлено, что в геноме третьим нуклеотидом кодона детерминирован один из трёх ротамерных вариантов пептидной связи, которым аминокислотный остаток (закодированный дуплетом первых двух нуклеотидом кодона) присоединяется к растущей полипептидной цепи (табл. 1).
Таблица 1. Генетический код структурного шаблона белка

XYZ – первый, второй и третий нуклеотиды в кодоне; R, 0, L – ротамерные варианты пептидной связи (РВПС); uaa, uag, uga – Stop-кодоны.
Ротамерные варианты пептидной связи различаются между собой углом поворота по оси пептидной связи ?, кратным 120о. Крайне важно понимать, что ротамерный вариант пептидной связи реализуется в процессе синтеза белка в рибосоме и дальнейшее вращение по уже образовавшейся полуторной пептидной связи становиться невозможным. Поэтому с рибосомы сходит совершенно индивидуальный структурный шаблон белка из последовательности ротамерных вариантов пептидной связи, в соответствие с информацией, содержащейся в его гене. Именно этим обстоятельством мы объясняем невозможность синтезировать нематричным способом функционально активные большие молекулы белковых ферментов или рецепторов. Твердофазный синтез реализован только для небольших неструктурированных пептидов, которые характеризуются избыточной конформационной подвижностью, в силу чего их функциональные конформации определяются взаимодействием с белками-партнёрами в составе гетерокомплексов, а не индивидуальным структурным шаблоном [13, с. 38].
Для конформеров вторичной структуры белка характерна периодичность, поэтому, правая спираль в структурном шаблоне белка кодируется последовательностью кодонов с С/G в третьей позиции, ?-тяж – повторением кодонов с А, а левая спираль – последовательностью кодонов с Т в третьем положении (табл. 2). Неструктурированные фрагменты кодируются чередованием кодонов с С/G, А и Т в третьей позиции.
Таблица 2. Кодирование конформеров вторичной структуры белка

У R, 0 и L-ротамеров все атомы пептидной группы (C? (i), C (i), O (i), N (i+1) H (i+1)) компланарны, кроме C? (i+1). C? (i+1) атом каждого аминокислотного остатка не принадлежит плоскости пептидной группы, благодаря чему происходит сворачивание полипептидной цепи в конформеры вторичной структуры белка (правая ?-спираль, ?-тяж, левая 3/10-спираль) ещё в рибосоме (рис. 2), а не после синтеза полипептидной цепи из практически единственного транс-изомера (цис-изомер только у пролина) в виде плоской ленты, сворачивание которой достигается поворотами на углы ? и ? по связям СО—C? и NH—C?, как предполагали ранее [9, с. 134]. Пластичность структурного шаблона в ходе посттрансляционного фолдинга и конформационная подвижность белка при взаимодействии с лигандами достигаются единственно возможным поворотом по оси связи NH—C? на приращение угла ?, кратное 120о [5, с. 139].

Рис. 2. Схема образования правого (R), нулевого (0) и левого (L) ротамерных вариантов пептидной связи.
Данный механизм трансляции генетической информации является эволюционно новым. Его формирование у эукариот было обусловлено необходимостью синтеза больших и сложных белков в виде структурного шаблона, максимально приближенного к функциональной конформации этих белков, чтобы их фолдинг имел наибольшие скорость и КПД. У прокариот и органелл эукариот (митохондрии, хлоропласты) третий нуклеотид кодонов в генах небольших полипептидов ещё не является информационным, поэтому на нём и наблюдается воблирование по описанному Ф. Криком механизму [12, с. 368]. Это обусловлено отсутствием пула изоакцепторных тРНК с модифицированными нуклеотидами в первом положении антикодона [6, с. 11] и нередко отсутствием филогенетически более молодых областей в структуре тРНК [15, с. 6730], т.е. недоразвитием звена, реализующего информацию третьего нуклеотида.
Выше изложенные положения легли в основу алгоритма компьютерных программ Secondary Structure Protein (SSP) и Three-dimension Structure Protein (TSP), которые по нуклеотидной последовательности мРНК позволяют смоделировать схему вторичной структуры и визуализировать индивидуальный структурный шаблон любого белка. Эту первичную информацию о белке можно использовать в дальнейшем моделировании фолдинга функциональной конформации белка с учетом физ-химии его микроокружения, посттрансляционных модификаций, взаимодействия с лигандами методами молекулярной динамики наравне с информацией о наиболее стабильном конформере, которую извлекают из рентгенограмм кристаллов белков. Преимущество данного подхода состоит в возможности быстрого моделирования индивидуальной пространственной структуры отдельной молекулы любого белка (даже если он не кристаллизуется, и никогда не сворачивается, как, например, регулятор клеточного деления Sic1 [11, с. 152]) с точностью до электрона (пикотехнология), опираясь лишь на информацию о нём в геноме. То есть, мы in silico воспроизводим трансляцию генетической информации в индивидуальный структурный шаблон белка, а не занимаемся поиском самой стабильной или «быстро достигаемой» устойчивой его конформации из 10100 возможных [14, с. 44], как это происходит при конформационном анализе поверхности потенциальной энергии молекулы белка громоздкими методами систематического поиска, Монте-Карло или молекулярной динамики с целым рядом ограничений и приближений [10, с. 41]. Не исключено, что большинство белков именно из конформации своего структурного шаблона максимально быстро, а главное однозначно, фолдируют в нативную конформацию с минимумом свободной энергии, формируя, таким образом, «устойчивое большинство» конформационно лабильного белкового пула.

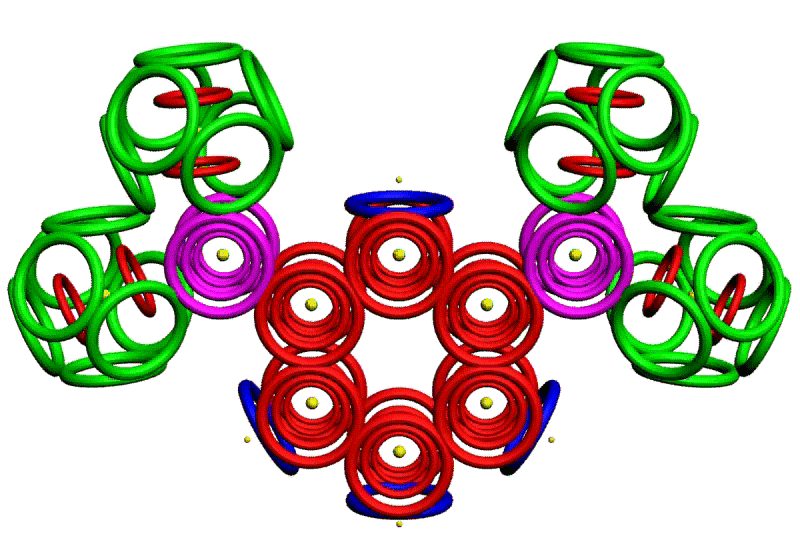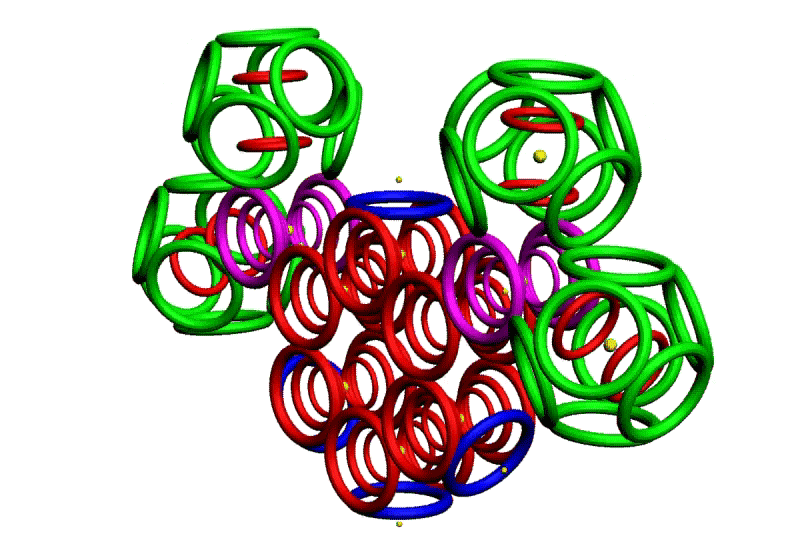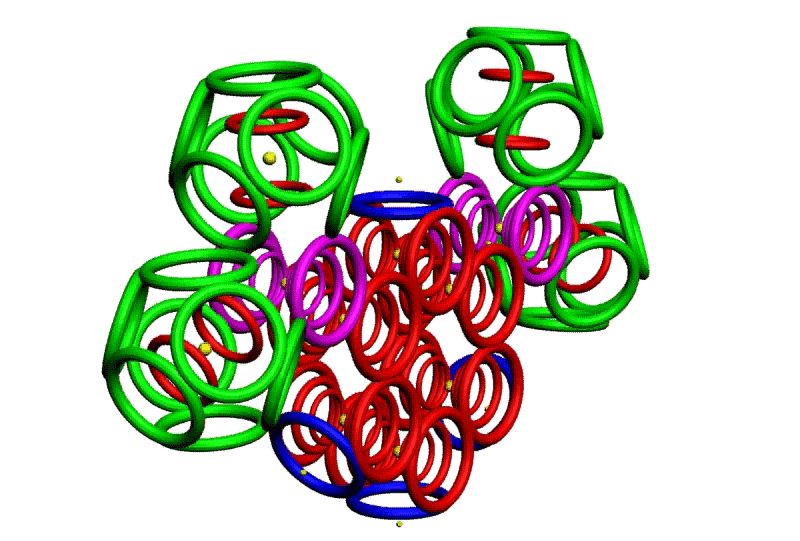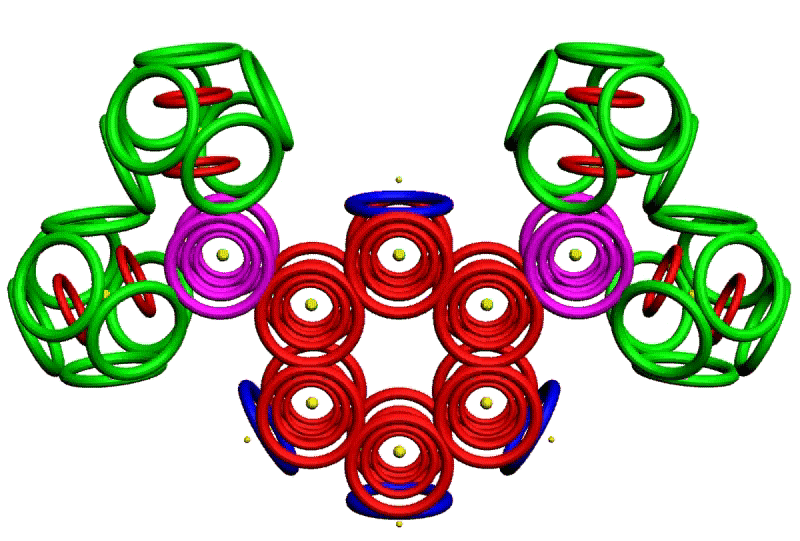
Рис. 3. Пикотехнологические модели структурных шаблонов некоторых белков и их комплексов.
В последнее время А.С. Спирин, отклоняясь от постулата о матричном синтезе белка в виде развёрнутой полипептидной цепочки [7, с. 5], предположил, что в самой рибосоме полипептид синтезируется сразу в виде ?-спирали и по желобу выталкивается наружу по мере трансляции мРНК [8, с. 437]. Этой прогрессивной гипотезе, которая основывается на подавляющем (74%) большинстве «спиральных» кодонов в генах эукариот, остался один шаг до представления о трансляции структурного шаблона белка не только в виде ?-спирали.
С помощью пикотехнологии были декодированы и смоделированы структурные шаблоны более 100 белков (рис. 3), сопоставительный анализ которых с экспериментальными данными Protein Data Bank (PDB) позволил подтвердить предположение о генетически закодированных размерах и местоположении конформеров вторичной структуры в нативной конформации этих белков [18, c. 348].
Итак, пикотехнология – это современный, точный и удобный методологический подход в арсенале молекулярной биологии для моделирования пространственной структуры белков, исходя из той информации генома о них, которой располагает сама клетка.
Список литературы
1. Кушелев А., Полищук С., Писаржевский С. Формы, механизмы, энергия наномира: Доступна ли энергия эфира для космических полётов? // Электроника: Наука, Технология, Бизнес. – 2002. – № 6. – С.72–76.
2. Кушелев А.Ю., Полищук С.Е., Неделько Е.В. и др. Построение масштабной модели структуры белка // Актуальные проблемы современной науки. – 2002. – № 2. – С. 236–243.
3. Нанонаука и нанотехнологии: энциклопедия систем жизнеобеспечения / Моск. гос. техн. ун-т им. Н. Э. Баумана; ред. О. О. Аваделькарим; гл. ред.: Чуньли Бай, С. П. Капица. — М.: Магистр-Пресс : Изд-во ЮНЕСКО : EOLSS, 2009. — 991 с.
4. Огжевальский З.И. 1972. Пространственные модели атомов, молекул и кристаллов. Москва, 1972. – 118 c.
5. Соколик В.В. Карта Рамачандрана: ротамерия пептидной связи и фолдинг белка // VII Международная научно-техническая конференция «Актуальный вопросы биологической физики и химии». БФФХ-2011, Севастополь. – 2011. – С.137–139.
6. Соколик В.В. Загадка изоакцепторных тРНК // II Всероссийская Интернет-Конференция «Актуальные проблемы биохимии и бионанотехнологии», Казань. – 2011. – С. 11-15.
7. Спирин А.С. Биосинтез белка: элонгация полипептида и терминация трансляции // Соросовский образовательный журнал. – 1999. – № 6. – С. 2–7.
8. Спирин А.С. Молекулярная биология: рибосомы и синтез белка. – М: «Академия», 2011. – 496 с.
9. Финкельштейн А.В., Птицын О.Б. Физика белка. - М.: Книжный дом «Университет», 2002. – 298 с.
10. Хёльтье Х.-Д., Зиппль В., Роньян Д., Фолькерс Г. Молекулярное моделирование. М.: БИНОМ. Лаборатория знаний, 2010. – 320 с.
11. Chouard T. Structural biology: Breaking the protein rules // Nature. – 2011. – V. 471, № 7337. – P. 151–153.
12. Crick F.H.C. The Origin of the Genetic Code // J. Mol. Biol. – 1968. – V. 38. – P. 367–379.
13. Fink A.L. Natively unfolded proteins // Curr. Opin. Struct. Biol. – 2005. – V.14, № 1. – P. 35-41.
14. Levinthal C. Are there pathways for protein folding // J. Chim. Phys. – 1968. – V. 65. – P. 44–45.
15. Maizels N., Weiner A.M. Phylogeny from function: Evidence from the molecular fossil record that tRNA originated in replication, not translation. // Proc.Nat.Acad.Sci.USA. – 1994. – V. 91, № 15. – P. 6729–6734.
16. Ratner M., Ratner D. Nanotechnology: a gentle introduction to the next big idea, 2003. – 195 p.
17. Snelson K. A design for the atom // Industrial design. – 1963. – № 1. – P. 48–57.
18. Sokolik V.V. Protein is coded in genome and synthesized in ribosomes as a structural template of a rotameric version sequence of peptide bound configuration // The International Moscow Conference on Computational Molecular Biology, МССМВ-11, Moscow. – 2011. – P. 347–348.
19. Sokolik V.V. Modeling of the individual structural template of protein on determining it nucleotide sequences // VII Международная конференция по биоинформатике, регуляции и структуры геномов и системной биологии. BGRS\SB-2010, Новосибирск. – 2010. – С. 275.
20. Sokolik V.V. Algorithm of protein structural template decoding according to its determined nucleotide sequence // Fist International Conference “Fundamental medicine: From scalpel toward Genome, Proteome and Lipidome”, Pax Grid Virtual Conferences, Kazan. – 2011. – P. 117–119.
Подписи под иллюстрациями.
Рис. 3: Гистоновый комплекс 5 H2A + Н2В_DROME (P84051, P02283)
Шаперон A0KFQ4_AERHH (A0KFQ4)
Суперспираль коллагена CO2A1_HUMAN (Р02458)
Белковый комплекс ?-протеина с ?-тубулином TAU_HUMAN + TBA1A_HUMAN
Инсулин INS_HUMAN (Р01308)
Лизоцим LYSC_CHICK (P00698)
Рибонуклеаза Н1 RNH_ECOLI (P0A7Y4)
Аполипопротеин Е APOE_HUMAN (P02649)
Гемоглобин (А-цепь) HBA_HUMAN (P69905)